

Research Projects
Introduction
The organization of DNA into dense chromatin structures presents an obstacle to the transcriptional machinery. Yet, it also provides an opportunity to regulate gene expression because chromatin structures can be locally and globally modified, making them highly dynamic. Structural changes in chromatin can be induced by the post-translational modification of histones or by the replacement of canonical histones with histone variants. While there is extensive knowledge of the enzymes that add or remove specific histone modifications or replace canonical histones with histone variants, it is not well understood in any organism how these enzymes are targeted to specific genomic loci. Furthermore, it is also not clear how transcriptionally active or repressed chromatin structures are established across the nucleus.
Thus, using trypanosomes to study the formation of chromatin structures, we hope to understand some of the evolutionarily conserved mechanisms of chromatin formation, while at the same time elucidating the role of epigenetic regulation in antigenic variation. Antigenic variation represents the key strategy used by the parasite to evade the host immune response.
Key questions of our research are:
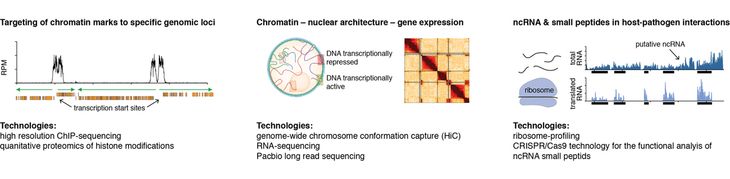
· How are histone variants and histone modifications targeted to specific genomic loci?
· How are chromatin, 3D genome architecture and gene expression linked?
· What role does regulatory ncRNA play in the regulation of gene expression?
Research Interests
We are currently investigating how, in the absence of specific DNA sequence motifs in T. brucei, the different histone modifications and histone variants are targeted to specific loci along the genome and what role they play in forming transcriptionally active or repressed chromatin regions across the nucleus.
To this end we have developed protocols that allow us to isolate chromatin from specific genomic loci. Furthermore, to analyze the chromatin, we established in collaboration with the group of Andreas Schlosser (University of Würzburg) a mass-spectrometry-based method allowing the precise quantification of histone modification levels (ElBashir et al. 2015, Analytical Chemistry). Together, these methods have allowed us to quantify the complete T. brucei histone acetylome, providing evidence for numerous novel acetyl and methyl marks. Furthermore, we revealed a strong hyperacetylation of nucleosomes at TSSs and identified at least one of the histone acetyltransferases responsible for this hyperacetylation (manuscript in preparation).
In addition to understanding how chromatin structures are established in 2 dimensions, we are interested in understanding the links between chromatin and the 3-dimensional genome architecture. The recent development of high-throughput sequencing-based assays to probe 3D genome architecture (Hi-C) has revealed the importance of the spatial organization of DNA inside the nucleus. For example, it was shown that 3D genome architecture plays a critical role in the regulation of mutually exclusive gene expression and the frequency of translocation between different genomic loci in many eukaryotes. Thus, we suspect the genome architecture to be a key regulator of antigenic variation. Yet, the causal links between genome architecture and the expression of antigens have not been studied systematically in any organism.
Recently, my group has established Hi-C and CRISPR-Cas9 technology in T. brucei (manuscripts in preparation). The establishment of these technologies has opened unprecedented opportunities to study the influence of DNA sequence elements on the spatial organization of DNA and the formation of chromatin structures. Our goal is to understand the links between chromatin, 3D genome architecture and antigenic variation.
Beyond chromatin dynamics, my group is interested in post-transcriptional mechanisms of gene regulation. The organization of genes in long polycistronic transcription units allows for very little regulation at the level of transcription initiation. Instead the unusual genome organization of trypanosomes points to an important role of post-transcriptional mechanisms in gene regulation, e.g. RNA maturation or the regulation of translation. However, the relative contribution of these processes to the final steady state levels of proteins is not well understood. To obtain genome-wide information on protein synthesis, we adapted a ribosome profiling approach to T. brucei. This technique is based on high-throughput sequencing of ribosome-protected RNA fragments and enabled us to perform the first genome-wide analysis of RNA translation and translational efficiency in a eukaryotic pathogen (Vasquez et al. 2014, Nucleic Acids Research).