Maria Colomé-Tatché

Computational epigenomics
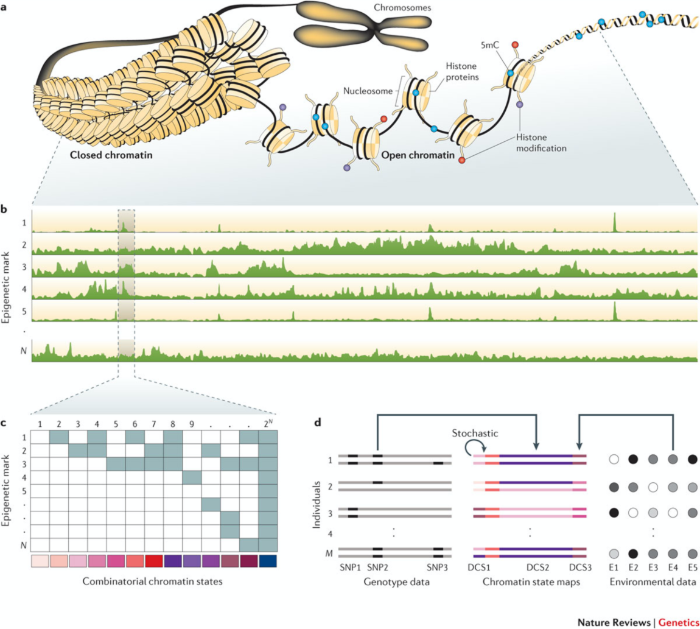
All cells in our body have the same DNA sequence; yet, we consist of hundreds of distinct cell types. It is becoming increasingly clear that the epigenome, a layer of chemical modifications to the DNA and its associated proteins, is primarily responsible for these differences as it regulates which genes are turned on or off in a cell-type specific manner. Understanding how the epigenome determines cellular identity requires genome-wide measurements of its molecular components, most notably DNA methylation, histone modifications and open chromatin. In the past decade, technological advances have enabled high-resolution measurements of these various epigenetic marks at the genome-wide scale, leading to the construction of so-called reference chromatin state maps (epigenomes) for different species and cell types.
Moreover, in the recent years epigenetic single cell measurements, where the epigenetic status of single cells is evaluated using next generation sequencing techniques, are becoming mainstream. Currently, two single cell epigenetic measurements are performed routinely in the lab: DNA methylation status can be assessed at the single cell level with the use of single cell bisulfite sequencing (scBS-seq), and open chromatin patterns are investigated at individual cells using single cell ATAC-seq (scATAC-seq). These measurements can be combined with the reading of the single cell transcriptome, either in the same cell or in the same cell population. Because of the possibility of linking transcriptomic and epigenetic states of the same individuals cells, these single cell measurements have the potential to answer many open biological and biomedical questions. However, analysing single cell epigenomic and transcriptomic data is challenging due the large data size, the high dimensionality of the data, the low signal to noise ratio and the extensive missing data.
In my group, we are interested in understanding how different epigenomes emerge, how stable epigenetic changes are, and how they lead to different phenotypes. In order to answer these questions, we develop computational methods and mathematical models to determine what regions of the epigenome are altered under different conditions in large numbers of individuals or in single cells, and how these changes affect the observed phenotype.