

Research Projects
Single cell computational epigenomics
We work on the development of computational models for the analysis of single cell DNA methylation data (i.e. single cell bisulfite sequencing) and single cell open chromatin data (i.e. scATAC-seq), and integration with single cell transcriptomics data. Integration is done using different techniques for scenarios where the different -omic layers have been measured for the same cell, or for matched cell populations. We are also interested in the calling of copy number variations (CNVs) from single cell data using different modalities. We have developed an R package for the calling of CNVs from single cell genomics data (aneufinder (https://bioconductor.org/packages/release/bioc/html/AneuFinder.html)) and a python package for the analysis of single cell DNA methylation data and single cell open chromatin data (performing common clustering, dimension reduction, trajectory learning, data integration and differential calling) (epiScanpy (https://episcanpy.readthedocs.io/en/latest/)).
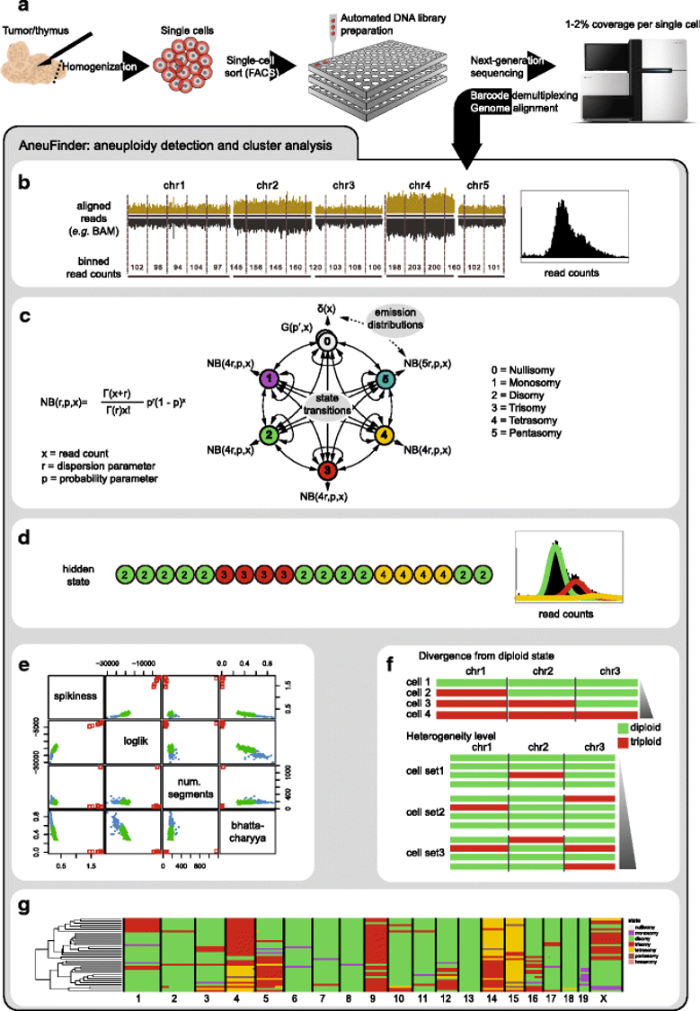
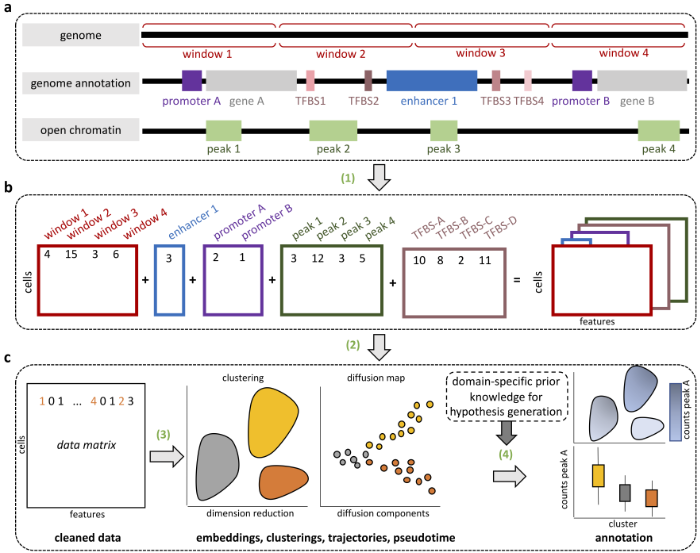
Computational epigenomics
We work on the development of improved analysis tools for the integration of several epigenetic marks, like DNA methylation and several histone modifications or chromatin openness. We are mainly interested in integrating large number of measurements in the same sample, or in situations where an epigenetic mark has been measured in a large number of samples or time points. We have developed an R package for the analysis of a large number of histone modifiations marks measured in several individuals or time points (chromstaR (https://bioconductor.org/packages/release/bioc/html/chromstaR.html)) and an R package for the imputation of DNA methylation levels and states in bisulfite sequencing data (METHimpute (https://bioconductor.org/packages/release/bioc/html/methimpute.html)).
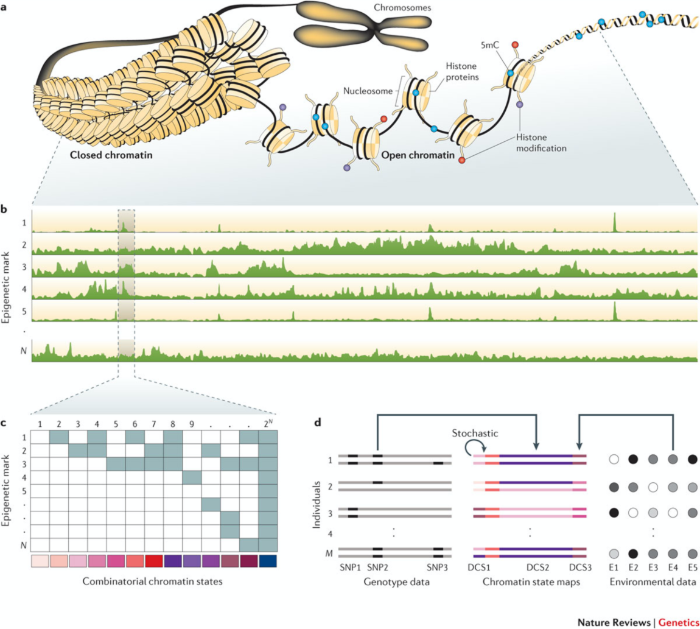
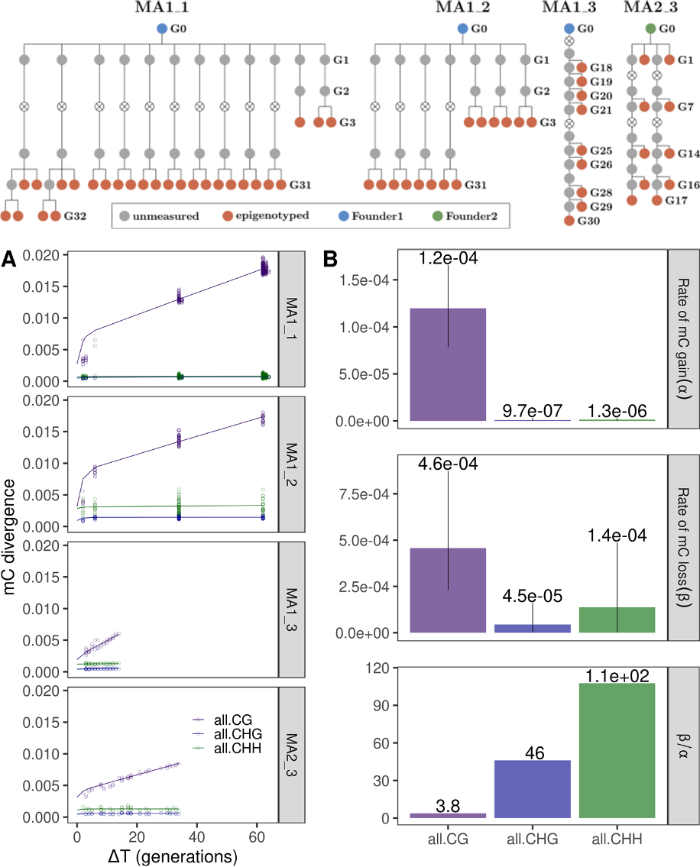
Mathematical modelling of epigenetic processes
We are interested in the modelling of dynamic epigenetic processes, such as DNA methylation inheritance. We are interested in epigenetic inheritance in plants, where it is well established that changes in DNA methylation can be transmitted across generations via meiotic inheritance. We work with inheritance of the epigenetic state of single cytosines as well as groups of cytosines (DMRs) with the aim of studying transgenerational as well as somatic epimutation rates. We are also interested in the inheritance of epigenetic states in mitotic cell divisions such as during embryonic development in mammals.
Finally, we work in modelling epigenetic fitness landscapes, to study the development of phenotypic advantage upon stress (like for example during treatment in the case of cancer development, or during immune response in the case of pathogen infection).
Biological and biomedical applications and collaborations
We work on different biological and medical applications, in strong collaboration with experimental partners. Our goal is to apply our developed methods to the analysis and interpretation of different sorts of single-cell and/or bulk multi-omic data. Our applications are mainly in the fields of cancer development and relapse, ageing, development and reprogramming, as well as in transgenerational epigenetic inheritance in plant populations.
We are part of the Marie Curie Innovative Training Network (ITN) “Cell to cell heterogeneity: What makes a successful pathogen: Understanding the impact of cell-to-cell heterogeneity in chromatin structure on infection and adaptation” (https://cell2cell.eu/) and of the Impuls und Vernetzungsfonds of the Helmholtz-Gemeinschaft Incubator grant “sparse2big” for the development of imputation methods in single cell data. We are a participating group in the graduate school “CausalCellDynamics”, which is an international partnership with the "Mila - Quebec AI Institute”. Maria Colomé-Tatché is a Principal Investigator in the Munich School for Data Science (MUDS) (https://www.mu-ds.de/) and an associate member of the SFB 1064 Chromatin Dynamics (https://www.sfb1064.med.uni-muenchen.de/index.html).